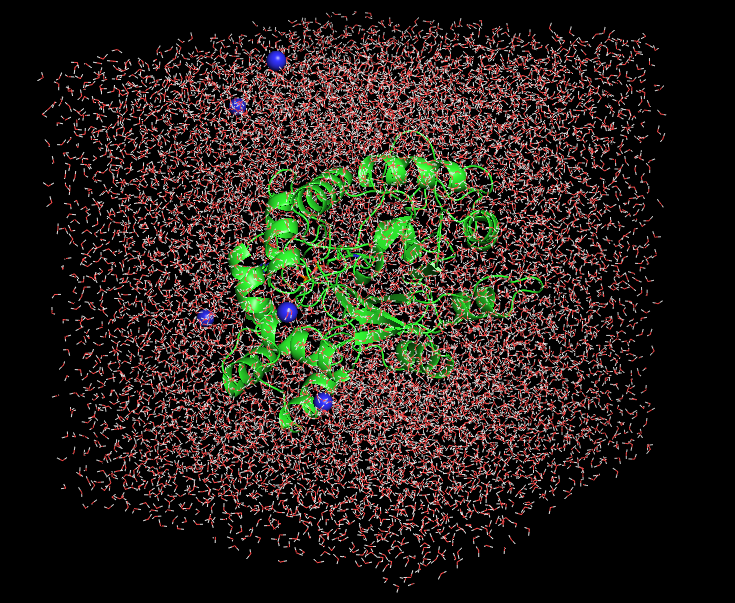
Molecular dynamics simulations (MD simulations) is one of the important tool in drug discovery to understand the stability of biological macro molecules and inhibitor complexes. Molecular Dynamics simulations (MD simulations) estimate time dependent behavior of a macro molecular system. Understanding the protein-ligand complex stability and the protein-protein interaction studies can also be studied by molecular dynamics simulations.
These MD simulations also determine the various properties of inhibitor molecules including their conformational change and fluctuations along with their biological macromolecules. At present molecular docking and Molecular dynamics simulations methods are widely used to explore in drug discovery and to understand thermodynamics of biological macromolecules.
MOLECULAR DYNAMICS SIMULATIONS COURSE
Software: GROMACS
OS: UBUNTU / WINDOWS10
COURSE CONTENET:
Introduction to molecular dynamics simulations
Introduction to GROMACS
Force field concept and types
Non-bonded interactions
Energy minimization methods
Solvation & periodic boundary conditions
Integration Algorithms
Molecular dynamics simulations and Results Analysis
Free energy calculations (MMPBSA/MMGBSA) and examples and hands-on.